|
Hepatotoxicity of inhalational agents 1. Types of Hepatic Injury A. Hepatocellular damage 1. Direct (obligate) hepatotoxicity, e.g. Chloroform, Carbon Tetrachloride.
2. Indirect (Facultative) or Immune-mediated hypersensitivity.
B. Interference with bilirubin metabolism C. Cholestasis D. Pre-existing liver disease or predisposing factors 2. Factors involved in hepatic injury A. Predisposing factors
B. Role of Biotransformation Once the direct hepatic toxicity of chloroform was shown to be due to its metabolism, interest in the metabolism of volatile anaesthetics has been considerable. Biotransformation is common for fat soluble unionised molecules, because they have generally longer half-lives (relatively more presented to liver) and readily diffuse into the endoplasmic reticulum within the cell (a hydrophobic region) for metabolism. For a given number of MAC hours exposure a higher proportion of the total amount inhaled is metabolised if the inspired concentration is low (and duration of exposure long). This is termed concentration dependence. Biostability of the molecule is obviously important. Stability of the C-H bond is enhanced by halogenation (fluorine > chlorine > bromine > iodine). Inferences about likely possible means of metabolism have been made from estimates of bond stability. C. Phases of Drug metabolism: Phase 1: Oxidation, reduction, hydrolysis (only for esters and amides). Cytochrome P450 is is a typical mixed-function oxidase, an iron containing enzyme which enhances the rate of oxidisation of many lipophilic drugs and is itself oxidised in the process. Elemental oxygen is required. It is "reactivated" by cytochrome P450 reductase. Carbon Monoxide can effectively inactivate this enzyme as it has 66 times greater affinity than oxygen for the oxygen-binding site. Initially only oxidation was thought to occur, but reductive metabolism of halothane has been shown to occur during ordinary clinical anaesthesia [1]. Phase 2: Conjugation Formation of water soluble forms allowing both excretion and enhanced rate of phase 1 reactions by removing the products from equilibrium. D. Possible mechanisms of toxicity involving biotransformation: Formation of inherently toxic breakdown products or intermediaries under normal conditions - risk is dose related (accumulation) - ie Fluoride from MOF, Chloroform. Formation of haptens with or without biotransformation, followed by immune-mediated hepatic destruction - e.g. possibly halothane. Free radical formation has been postulated to be the mechanism of microsomal damage where enzyme function is involved. These ionised molecules remove hydrogen atoms from fatty acids resulting in lipoperoxidisation and ultimately structural and functional changes in the mitochondrial endoplasmic reticulum. Free radicals are normally scavenged by the anti-oxidant glutathione. Where enzyme induction is required to cause toxicity, an at risk group influenced by age, genetics, enzyme induction (environment), etc may be present in the population. 3) CHLOROFORM While hepatitis following chloroform administration caused two deaths in its first year of use, it was more pleasant to use than ether and largely because of the efforts of Snow and Simpson became popular. It fell out of favour around the turn of the century, largely because of its tendency to cause respiratory depression and its potent enhancement of myocardial irritability, leading to sudden death due to VF, often soon after induction in healthy patients. 1847 - Chloroform first used for anaesthesia (Simpson) on suggestion of Waldie. 1850 - Caspar "delayed chloroform poisoning". 1894 - Guthrie reports series of cases of hepatotoxicity. 1912 - AMA Committee on Anaesthesia recommends discontinuation of its use. 1969 - Cohen & Hood using autoradiography in mice showed covalent binding of radioactive 14C confined to the liver following 14C-labelled chloroform anaesthesia. Subsequently the extent of covalent binding was shown to correspond to the degree of hepatic damage, and that both were increased by enzyme induction with agents such as phenobarbitone, and reduced by the enzyme-inhibitor, piperonyl butoxide. 1970 - Brown demonstrated increased toxicity associated with lipoperoxidation following enzyme induction with phenobarb and reduced toxicity following enzyme inhibition with disulfuram. He postulated the formation of the short-lived free radical *C Cl3 : C Cl2. 1980 - Pohl demonstrates formation of trichloroethanol by microsomal P450 which spontanously degrades to phosgene ( COCL2), which destroys glutathione and forms reactive Chloride. 1983 - Baden demonstrates carcinogenicity following chronic administration[2]. Hepatic damage occurs within 30 minutes of commencing chloroform anaesthesia, and increased bleeding due to reduced prothrombin production occurs within 1-2 hours. Severe hepatic damage due to chloroform has the following features:
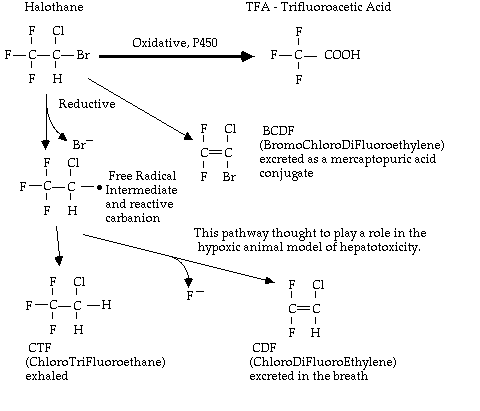
Metabolism (up to 50%): CHCl3 → CCl3OH → COCl2 (phosgene) → CO2 + Cl2 It seems that the cytochrome P450-mediated oxidative metabolism of chloroform results in the formation of inorganic chloride (excreted in the urine), CO2 (exhaled), phosgene, and some hepatic covalently bound carbon (either via free radical or phosgene formation). Metabolism has been demonstrated to essential for hepatic injury. While chloroform is not used these days, the hepatotoxic mechanisms involved are very relevant to the question of "halothane hepatitis". 2) HALOTHANE CF3-CHClBr History Over 900 cases of hepatitis after Halothane in the first 20 years of use. 1969 - National Halothane Study. Designed to determine if "Halothane Hepatitis" existed and if so, to determine its incidence. 865,000 cases, of which 250,000 used halothane 82 cases of fatal hepatic necrosis 9 episodes of "massive hepatic necrosis of unexplained aetiology" 7 of these followed Halothane, 4 on more than one occasion in the last 6 weeks Hence incidence 7 per 250,000 or 1:35,000. However, Dykes noted that not one patient who was jaundiced after halothane and died after a subsequent halothane anaesthetic was found at necropsy to have suffered massive or intermediate hepatic necrosis. No causal relationship between halothane and fatal postoperative hepatic failure found. Overall incidence of death with halothane similar to or less than other agents. Hence no resolution of the issue - only added fuel to the controversy. 1969 - Klatskin & Kimberg reported a positive challenge test in an anaesthetist [3] , although skeptics pointed out that this person had many episodes of hepatitis, not all of which occurred while exposed to halothane. 1970's - General acceptance in the USA that halothane hepatitis may exist. Studies to evaluate possible mechanisms commenced. 1976 - Walton et al comprehensively reviewed postoperative hepatic dysfunction from 5,000,000 anaesthetics (1/4 halothane) in the UK, and of 204 cases of postoperative jaundice a panel of hepatologists classified 76 as unexplained and following halothane. 95% of these followed multiple halothane anaesthetics, 55% within 4 weeks. An association between obesity and middle-aged women was found. 1976 - Rat mild hypoxia model following enzyme induction - Gandolfi & Van Dyke (phenobarbitone) [4] and Reynolds & Moslen (Polychlorinated biphenyls) [5]. Demonstrated animal model of massive hepatic injury following enzyme induction. Reductive metabolites were found to be covalently bound to hepatic microsomes (site of the reductive process). Aroclor 1254 pretreatment can cause centrilobular necrosis even without hypoxia in rats exposed to halothane. 1980 - Vergani et al reported that serum from patients who had suffered massive hepatic necrosis following halothane contained antibodies specific for isolated rabbit liver cell membranes which had been previously exposed to halothane [6]. Lymphocytes were shown to destroy the complex. Concept of halothane initiated hepatic autoimmunity. 1981 - Three pairs of closely related patients with halothane hepatitis reported by Hoft & Bunker [7]. 1982 - Shingu et al showed that hypoxia per se could induce a similar picture [8] [9]. 1982 - Bentley & Gandolfi show evidence of greater reductive metabolism in obese patients [10]. 1982 - Plummer & Cousins using NMR "spin trapping" demonstrated free radical formation in rats who developed hepatic injury after halothane and carbon tetrachloride but not after enflurane or isoflurane in an hypoxic animal model [11] 1983 - Neuberger et al studied 16 patients with hepatic failure following exposure to halothane within the previous month [12]. In 8 patients the halothane related antibody was detected, and of the others a firm diagnosis of viral hepatitis was made in 5, one demonstrated a hepatic auto-antibody, and the other two were unexplained. Clinical courses were identical whether or not the halothane associated autoantibody was detected. 1984 - Plummer et al found that reductive metabolism as measured by exhaled CTF and CDF was not reduced in childhood [13]. 1986 - Lunam & Cousins describe strain differences in halothane hepatotoxcity in guinea pig model [14] . Others have also demonstrated sex differences. Guinea pigs reliably get hepatic injury after 4h at 1%. 1994 - Lind & Gandolfi describe dose-dependent fatal halothane hepatotoxicity in a glutathione depleted guinea pig model. Due to covalent binding of reactive trifluoroacyl acid intermediates via oxidative CP450 pathways. Metabolism of Halothane
Approximately 20% of an inhaled dose is oxidatively metabolised (P450 mediated and phenobarbitone enhanced) to trifluoroacetic acid (TFA). Results in increased serum Bromine excreted in the urine which peaks at 1-2 days after exposure. A small amount is also metabolised by inducible enzymatic reductive pathways also using P450 but without oxygen (increased greatly below PO2 of about 10 mmHg in hepatic venous blood), and this generates free radicals capable of damaging microsomal structures in hepatocytes. Their presence has been documented by the presence of exhaled metabolites, urinary fluoride excretion, and free radical spin trapping. Several studies have shown greater increases in postoperative enzymatic tests of hepatocellular damage following halothane when compared to other agents, but these have not been reliably confirmed.
Note these exceed the 10 kCal /mol available for spontaneous degradation, hence enzymatic actions are required. Also notethat the C - Br bond is most susceptible. Mechanisms of Toxicity Metabolite-mediated direct toxicity Evidence for:
Against:
Immunologically-mediated damage to liver cells For:
Against:
Hypoxia alone For:
Against:
Something else?
Described features of halothane hepatitis [16]
Recommendations for use of halothane In view of likely increased risk of rare but fatal hepatotoxicity associated with the use of halothane as a volatile supplement to general anaesthesia when compared to other agents: Do not use after prior history or family history of unexplained jaundice after halothane? Do not use unless indicated? Cousins : "exercise care if obese, pre-existing liver disease, hypoxia likely", and avoid if several of the following are present: over 40 years old, long operation, biliary surgery, female patient, obese, allergic, drug dependent, sepsis, exposure less than 3 months previously. 2) ENFLURANE CHF2-O-CF2-CHClF By spring of 1983, 89 cases of hepatitis following enflurane anaesthesia had been reported in the US over the previous 10 years, and 10 were published. In 24 cases there was no ready explanation, of these 6 were severe and two resulted in death. 43 million enflurane anaesthetics were estimated to have been given during this period. The reported incidence of "enflurane hepatitis" fell during the study period. Hypoxic rat studies showed similar hepatic damage could be produced after enflurane if the inspired oxygen was reduced further (and also after fentanyl). Enflurane was shown to reduce hepatic blood flow less than halothane. Inorganic fluorine may rise to levels that may impair renal function only after very prolonged periods. 3) ISOFLURANE CHF2-O-CHCl-CF3 Hypoxic rat model showed hepatic damage was possible at a similar extent to enflurane. 4) NITROUS OXIDE MAC 105% One suggestion of a facilitatory hepatotoxic role [18], and nitrous oxide was used in virtually all cases of unexplained hepatitis. Possible role could involve increased risk of hypoxia, and inhibition of methionine synthetase. Note that uptake of Nitrous is quicker than desflurane, for two reasons, firstly the tissue/blood partition coefficients are a little higher, and secondly because or the concentration effect on induction with nitrous. 5) SEVOFLURANE CH2F-O-CH-(CF3)2 1st synthesised in the 1970's; delayed development because of possible toxicity. 1,000,000 anaesthetics given with this agent by end-1993 in Japan. Low pungency; suitable for inhalational induction. No similarities to other commercial agents, but like Desflurane is soley composed of C,F, and H. Can react with soda lime to form an olefin molecule called `Compound A' (CH2F-O-C(CF3)=CF2 ), which may cause organ toxicity and can reach signficant concentrations in a closed or very low flow circle circuit, and `Compound B' (CH2F-O-CH(CF3)-CF2-O-CH2F), which doesn't do much of either. Reaction is enhanced by temperature, varies according to type of soda lime (baralyme worse than soda lime), and is worse if the soda lime is dry. Compound A is lethal to rats at 400ppm and may cause mild tubular necrosis at 50ppm. Bito showed Compound A levels in humans exposed to 1% sevoflurane for 5hrs ranged from 12-40ppm with soda lime and 24-42ppm with baralyme. Keeping the soda lime refrigerated can reduce this a lot, bit nevertheless these levels are high. About 3% metabolised in the patient; ie, 100 times as much as desflurane. Cytochrome P450 2E1. Organic metabolite is hexafluororisopropanol glucuronide, excretion of which peaks at 12hrs with t½ of 55 hrs. 6) DESFLURANE CF2H-O-CFH-CF3 1st synthesised in the 1960's, but difficult and dangerous to produce, so went ahead instead with enflurane. Only structural difference is replacement of chlorine on the alpha ethyl carbon with fluorine. Irritant to inhale. Needs special electrically-heated vapouriser. Most rapid onset and offset of all volatile agents. Stable with CO2 absorber materials; can be used in closed circuit for long periods of time. Virtually no metabolism; no inorganic fluoride detectable above baseline after 7 MAC hrs. OTHER CAUSES OF POSTOPERATIVE HEPATIC DYSFUNCTION
REVIEW ARTICLES Mazze RI: Metabolism of the inhaled anaesthetics: implications of enzyme induction. Br J Anaesth 56 : 27s-44s, 1984. Stock JGL, Strunin L: Unexplained Hepatitis following halothane. Anaesthesiology 63: 424-439, 1985. Eger, EI: New Inhaled Anesthetics. (review article) Anesthesiology 80:906-922, 1994 REFERENCES 1. Sharp JH, Trudell JR, Cohen EN: Volatile metabolites and decomposition products of halothane in man. Anaesthesiology 50 : 2, 1979. 2. Baden JM: Chronic toxicity of inhalational anaesthetics. Clin Anaesthesiol 1:441, 1983. 3. Klatskin G, Kimberg DV: Recurrent Hepatitis attributable to halothane sensitisation in an anaesthetist. N Engl J Med 280: 515–512, 1969. 4. Widger LA, Gandolfi AJ, Van Dyke RA: Hypoxia and halothane metabolism in vivo: release of inorganic fluoride and halothane metabolite binding to cellular constituents. Anaesthesiology 44:197, 1976. 5. Reynolds ES, Moslen MT: Halothane hepatotoxicity: enhancement by polyclhorinated biphenyl pretreatment. Anaesthesiology 58: 237, 1977. 6. Vergani D, Vergani A, Alberti A: Antibodies to the surface of halothane altered rabbit hepatocytes in patients withj severe halothane associated hepatitis. N Engl J Med 303 : 66-71, 1980. 7. Hoft RH, Bunker JP, Goodman HI, Gregory PB: Halothane hepatits in three pairs of closely related women. N Engl J MEd 304: 1023-1024, 1981. 8. Shingu K, Eger EI 11, Johnson BH: Hypoxia per se can produce hepatic damage without death in rats. Anaesth Analg 61: 820-823, 1982. 9. Shingu K, Eger EI 11, Johnson BH: Hypoxia may be more important than reductive metabolism in halothane induced hepatic injury. Anaesth Analg 61: 824-827, 1982 10. Bentley JB, Vaughan RW, Gandolfi AJ, Cork RC: Halothane biotransformation in obese and non-obese patients. Anaesthesiology 57: 94-97, 1982. 11. Plummer et al: Free radical formation in vivo and Hepatotoxicity due to Anaesthesia with halothane. Anesthesiology 57: 160-166, 1982. 12. Neuberger J, Gimson AES, Davis M, Williams R: Specific serological markers in the diagnosis of fulminant hepatic failure associated with halothane anaesthesia. Br J Anaesth 55 : 15-18, 1983. 13. Plummer JL, Van Der Walt JH, Cousins MJ: Reductive metabolism of halothane in children. Aneasth Intens Care 12: 293-295, 1984. 14. Lunam CA, Cousins MJ, Hall PM: Genetic disposition to liver damage in a guinea pig model of halothane hepatotoxicity. Anesth Analg 1986? 15. Gelman S, Rimerman V, Fowler KC, Bishop SP, Bradley EL: The effect of Halothane, Isoflurane, and blood loss on hepatotoxicity and hepatic oxygen availability in phenobarbital-pretreated hypoxic rats. Anesth Analg 63: 965-972, 1984. 16. Neuberger J, Williams R: Halothane Anaesthesia and liver damage. Br Med J 289: 1136-1139, 1984. 17. Carrigan TW, Straughen WJ: A report of hepatic necrosis and death following isoflurane anaesthesia. Anaesthesiology 67: 581-583, 1987. 18. Ross JA, Monk SJ, Duffy SW: Effect of Nitrous Oxide on halothane induced hepatotoxicity in hypoxic enzyme induced rats. Br J Anaesth 56: 527-533, 1984.
Last updated Tuesday, December 15, 2020 |
|||||||||||||